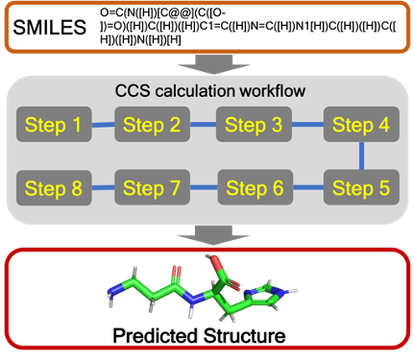
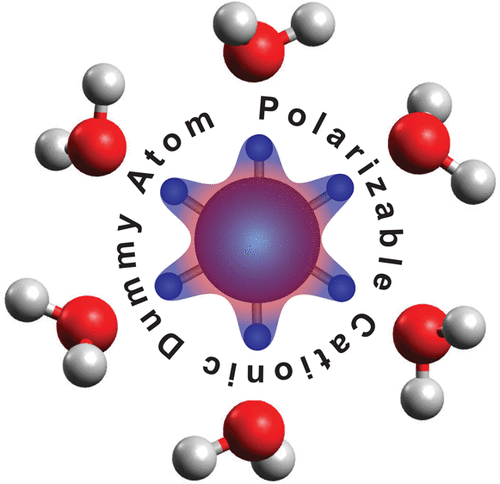
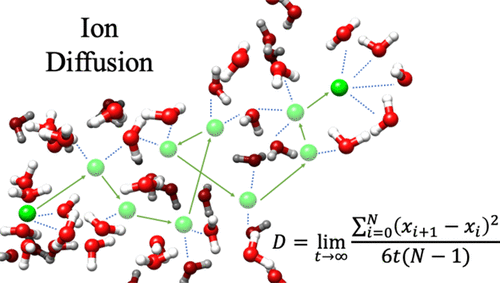
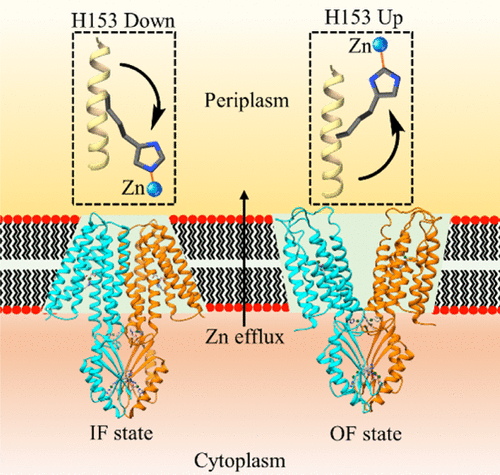
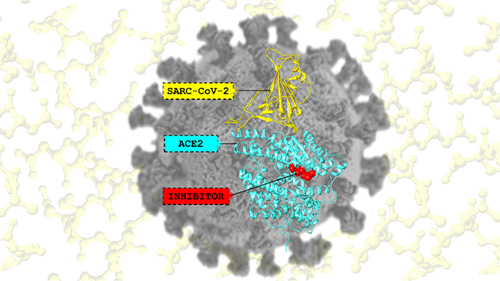
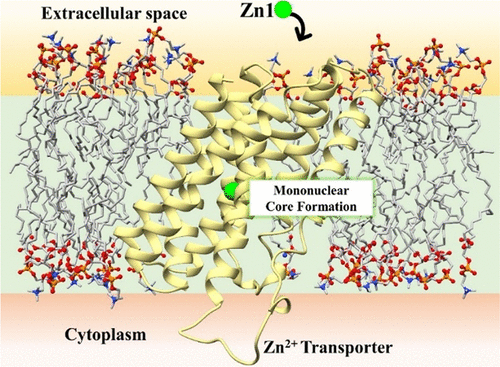
We are involved in research at the interface between the computational sciences and biology. We work on a number of problems and collaborate with experimentalists at every opportunity. Research areas of most interest include computer-aided drug design (CADD), using free energy methods to compute relative and absolute free energies of biological processes, metal ion force field design, metalloenzymes and metal ion homeostasis, development and application of quantum mechanical methods to biological problems and development of metabolomics workflows for NMR and CS computations. For further details go to the research section and look over our publications. Link on the images below for some of our recent publications.
|
The Portal for Open Computational Metabolomics Tools (POMICS) provides advanced computational methods to the metabolomics community.
|

|
|
|
|
|